back

Ginkgo × Biomni Lab: Biomni Democratizes Rapid Interpretation of Complex Transcriptomic and Cell Painting Data
We're excited to announce our partnership with Ginkgo Datapoints.
Jan 28, 2026
At Phylo, we’re building AI systems that help biologists turn complex, high-dimensional data into insight—faster and with far less friction. A major barrier in modern biology isn’t the lack of data, but the difficulty of interpreting it at scale. To showcase how natural-language–driven analysis can unlock value from rich functional genomics resources, we are excited to partner with Ginkgo Bioworks and applied Biomni Lab to their open-sourced GDPx datasets: GDPx1 and GDPx2 (chemical perturbations in primary cells profiled with DRUG-seq) and GDPx3 (chemical perturbations profiled with Cell Painting). These datasets represent some of the highest-quality public perturbation resources available, spanning diverse cell types, compounds, and doses.
Each GDPx dataset captures thousands of transcriptomic or morphological measurements per condition, offering an unusually detailed view of cellular responses across biological contexts. Yet extracting insight from data of this scale typically requires extensive coordination between specialists, long analysis cycles, and significant computational effort—leaving much of the signal inaccessible to bench scientists. This makes the GDPx collection an ideal proving ground for Biomni Lab: a virtual research environment designed to collapse analysis, interpretation, and visualization into a single conversational workflow.
More about Ginkgo Datapoints
At Ginkgo Bioworks, we're breaking down barriers to generate and use data effectively, accelerating research and advancing scientific discovery. We've open-sourced a series of functional genomics datasets designed to democratize access to high quality data: GDPx1 + GDPx2 (chemical perturbations in primary cells profiled with Drug-seq) and GDPx3 (chemical perturbations in primary cell profiled with Cell Painting). These resources provide researchers with the high-quality perturbation data needed to drive meaningful insights in biomedical research.
These datasets contain massive insights about cells across diverse conditions—cell types, treatments and doses. Each condition yields thousands of measurements from transcriptomics and high-content imaging. But most of the insights remain buried. This is because traditional analysis approaches have significant bottlenecks:
Coordination complexity: Multiple specialists across bioinformatics, cell biology, and pharmacology must work together to finish the task.
Time bottlenecks: Teams spend weeks to months on manual data processing, bioinformatics analysis, visualization, and hypothesis testing.
The result? Potential discoveries remain locked in data while scientists wait for computational resources and expertise to become available.
GDPx2 DRUG-seq Analysis
We asked Biomni to analyze disease signatures across 89 compounds from our GDPx2 DRUG-seq dataset. This comprehensive transcriptomic profiling dataset contains 85 LOPAC compounds tested across multiple concentrations in human primary cell types- Aortic Smooth Muscle Cells, Dermal Fibroblasts, Skeletal Muscle and Epithelial Melanocytes.
We generated two categories of prompts:
Starting from the raw counts data and metadata we first aimed to reproduce and visualize results we had obtained from our own bioinformatics analysis as shown in our BioRxiv preprint. Starting from raw counts we were able to re-generate Figures 2a,b,c and Supplementary Figure2 in the pre-print with specifying the analysis tool and associated parameters. We are not showing those results here.
For more complex tasks such as differential expression, pathway enrichment and dose-response calculations, instead of performing the bioinformatics tasks, we focused on interpretation and visualization of the processed data. In addition to the Raw Counts data and Metadata, we supplied the agent with processed data such as differential-expression, GSEA pathway-enrichment as well as dose-response data to Biomni. Biomni was able to synthesize large amounts of data and summarize it quickly providing areas to focus on. We found Biomni to be most helpful in this context. We were able to verify answers to specific questions that relied on the results of our processed data.
Some examples of prompts are listed below:
Question 1:
Using DoseResponse.csv in the data lake for GDPx2, plot a ranked list showing the number of dose-dependent-genes split by cell type and compound.
The number of dose-dependent genes observed by cell-type match our expectations as described in our BioRxiv preprint, with the highest number of dose-dependent genes attributed to our controls Trichostatin-A and Brefeldin-A.
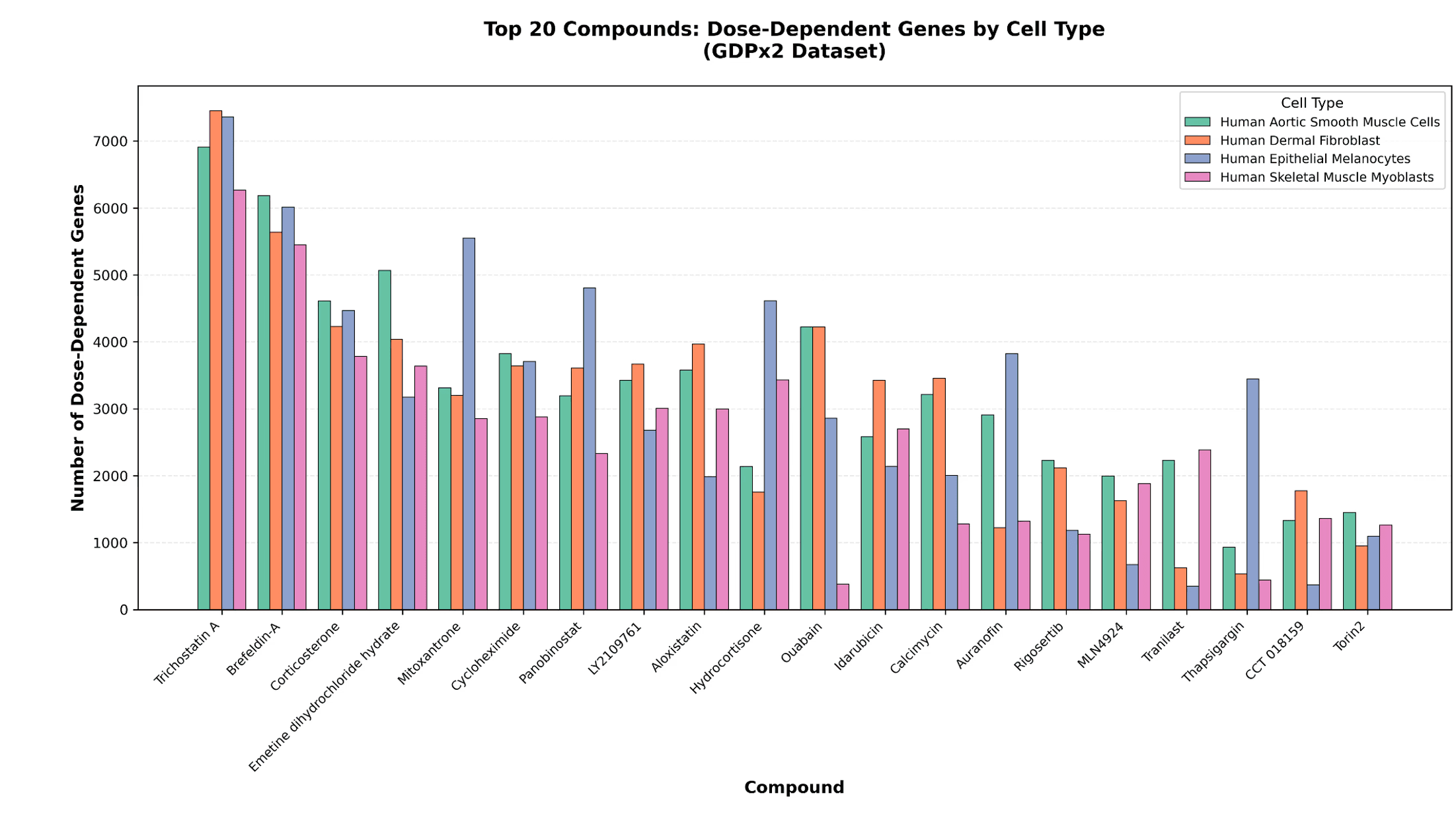
Figure 1. Dose-dependent gene numbers of top20 compounds by cell-type.
Question 2:
Using DoseResponse.csv in the datalake for GDPx2, which cell types have the highest number of dose-dependent genes in response to HDAC inhibitors TrichostatinA and Panobinostat?
In line with our expectations, Epithelial Melanocytes respond to HDAC inhibitors the strongest and have the highest number of common genes.
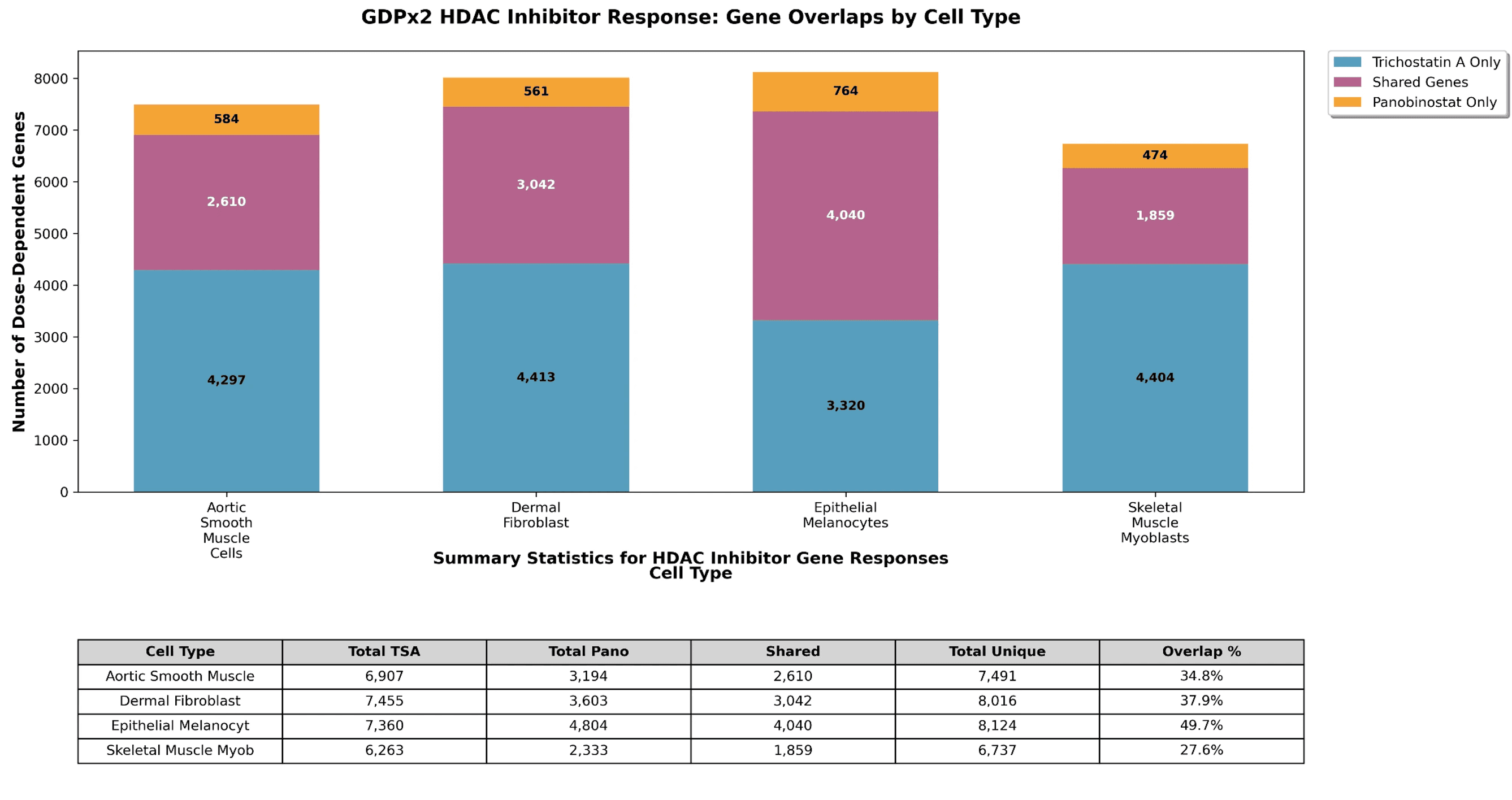
Figure 2. HDAC inhibitor dose-dependent gene response across cell types.
Question 3:
Using DoseResponse.csv in datalake for GDPx2, which cell type is most responsive to Dexamethasone? Which cell type is most responsive to dexamethasone- show number of DEGs and mean(abs(log2FC))?
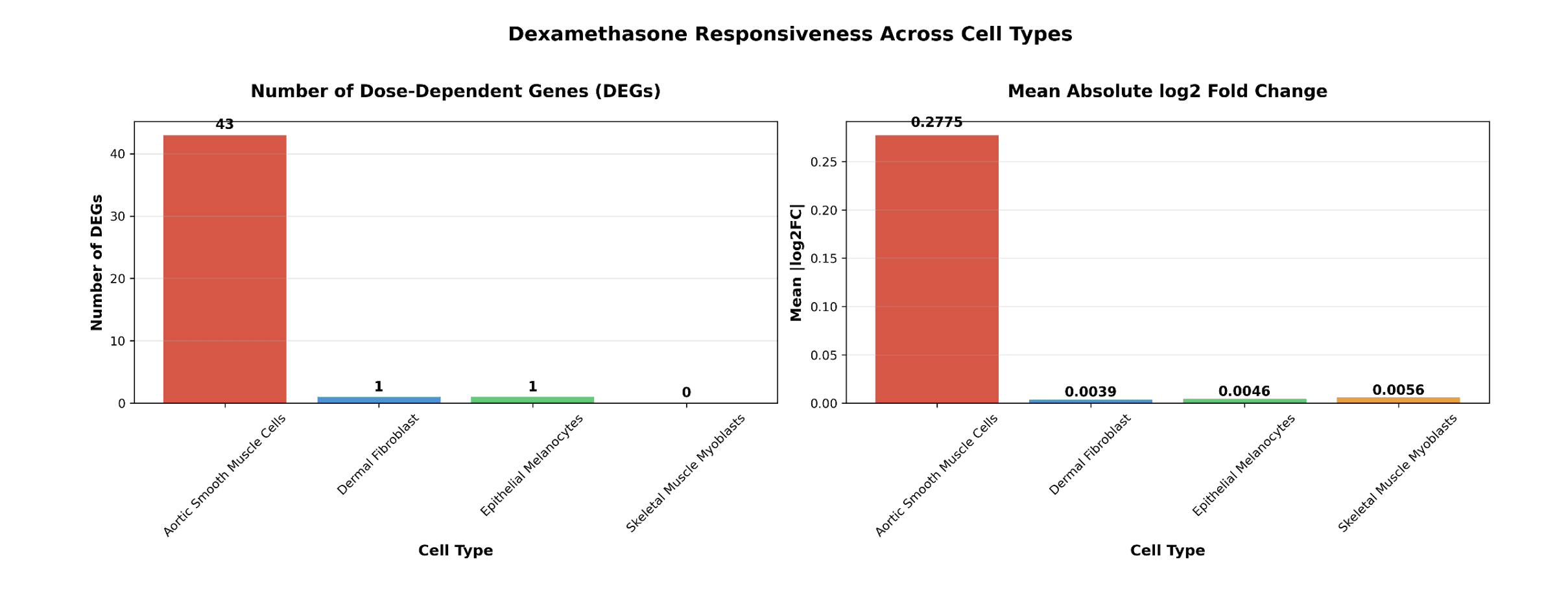
Figure 3. Dexamethasone cell-type specific responses.
As we expected the AoSMC’s have the largest amount of dose-dependent genes in response to dexamethasone and other glucocorticoid targeting drugs.
Question 4:
Using GDPx2_GSEA-HallmarkGenes.csv in datalake for GDPx2, which pathways do glucocorticoid drugs activate or inhibit in Aortic Smooth Muscle Cells? Include mean pathway effects across all glucocorticoid drugs.
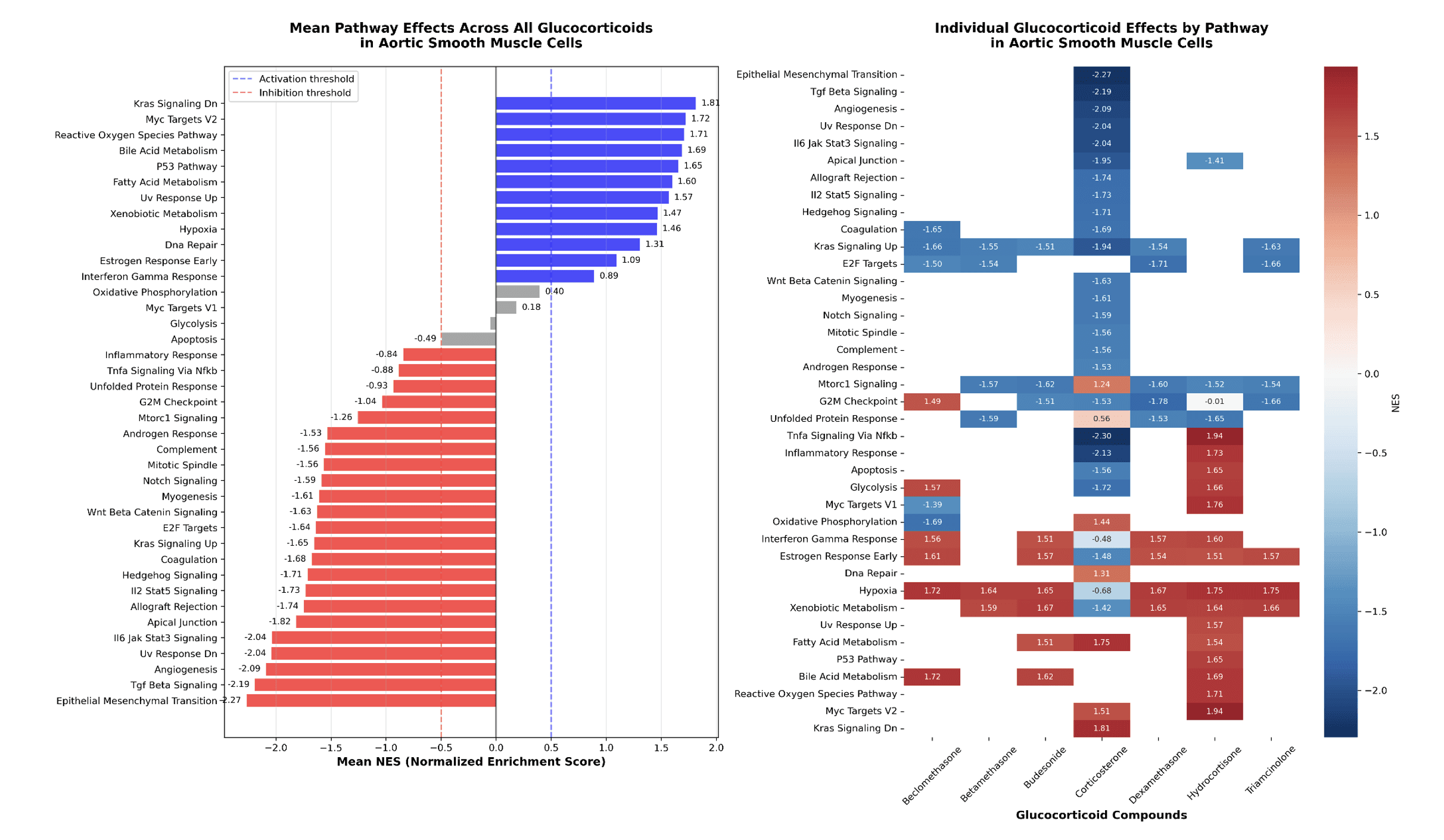
Figure 4. MSigDB Pathway Enrichment for glucocorticoid targeting drugs.
We expected to observe Hypoxia and Xenobiotic metabolism pathway genes to be enriched for all glucocorticoid targeting drugs. In addition, Corticosterone displayed a general downregulation of multiple pathways.
Question 5:
Using GDPx2_GSEA-HallmarkGenes.csv in the datalake, list the pathways that are related to cell-killing and identify compounds which lead to common cell-killing in all cell types and in at least 3 concentrations. Calculate mean NES for the 3 concentration with the highest significant NES and across the four cell types.
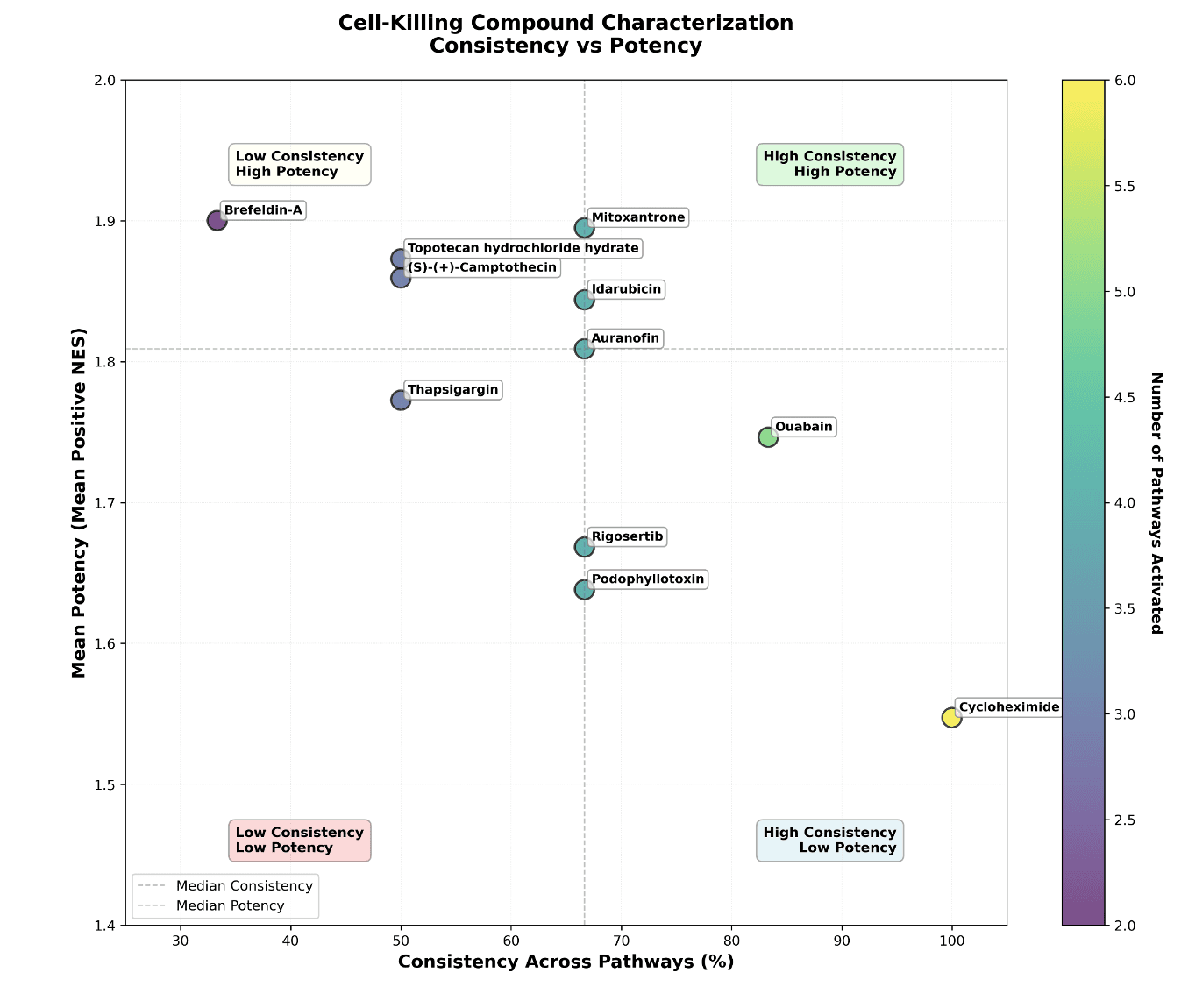
Consistency = (Number of pathways with positive NES) / 6
Mean Potency = Mean(all positive NES values for compound)
Most Consistent (Pathway Breadth):
Cycloheximide (100% - all 6 pathways)
Ouabain (83% - 5/6 pathways)
Multiple at 67% (4/6 pathways)
Most Potent (Mean NES):
Brefeldin-A (1.900)
Mitoxantrone (1.895)
Topotecan (1.873)
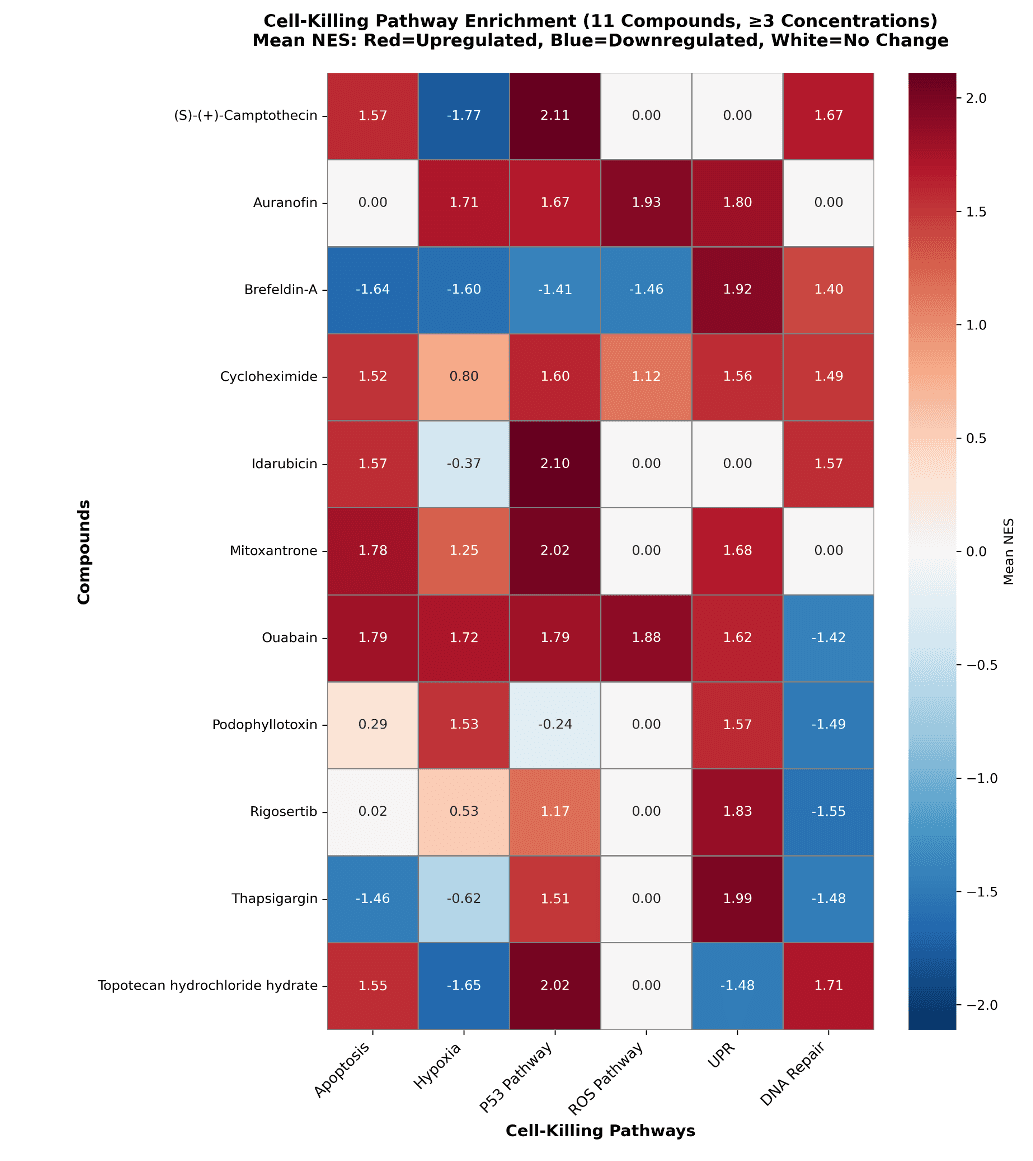
Figure 5. (A) Scatterplot of cross-pathway NES vs Mean NES scores. (B) Drugs with common cell-killing mechanisms across cell types.
Biomni identified 11 compounds that activate cell-killing pathways across all 4 cell types (human aortic smooth muscle cells, dermal fibroblasts, epithelial melanocytes, and skeletal muscle myoblasts) in at least 3 concentrations. was able to summarize and visualize cytotoxic drugs and the pathways leading to cell-death across the four cell types. It also suggested multiple forms of visualizations side-by-side making it easy for the analyst to decide on the best option. Drugs like Ouabain, Cycloheximide, Idarubicin, and Mitoxantrone are affecting multiple cell-killing pathways and with high potency.
GDPx3 Cell Painting Analysis
Similarly, we asked Biomni to analyze morphological features extracted from a CellPainting dataset of 45 LOPAC compounds tested across A549, Dermal Fibroblasts, Skeletal Muscle, Aortic Endothelial Cells. We asked Biomni to perform a step-by-step QC of provided data structures and provide comprehensive summaries and data visualization for CellProfiler extracted morphological features from Cell Painting.
Example prompts are listed below:
Question 6:
“I am analyzing data from a chemical perturbation dose response screen with cell painting readout performed in 4 cell types. I have feature data at GDPx3_median_selected.csv, metadata at GDPx3_metadata.csv, and metadata column information at Ginkgo GDPx3_metadata_columns.csv.
For each cell-type, zscore transform the columns in GDPx3_median_selected.csv values. For each cell-type generate a plot for top 20 PC’s for variance explained by timepoint, concentration, compound, container_id.
On a separate plot, generate a principal component analysis of the features plot PC1 vs PC2 with timepoints (24 and 48 hrs) colored in one set of plots and colored by sample type in another set of plots. Include a UMAP visualization using the top 20 PCs colored by sample type.”
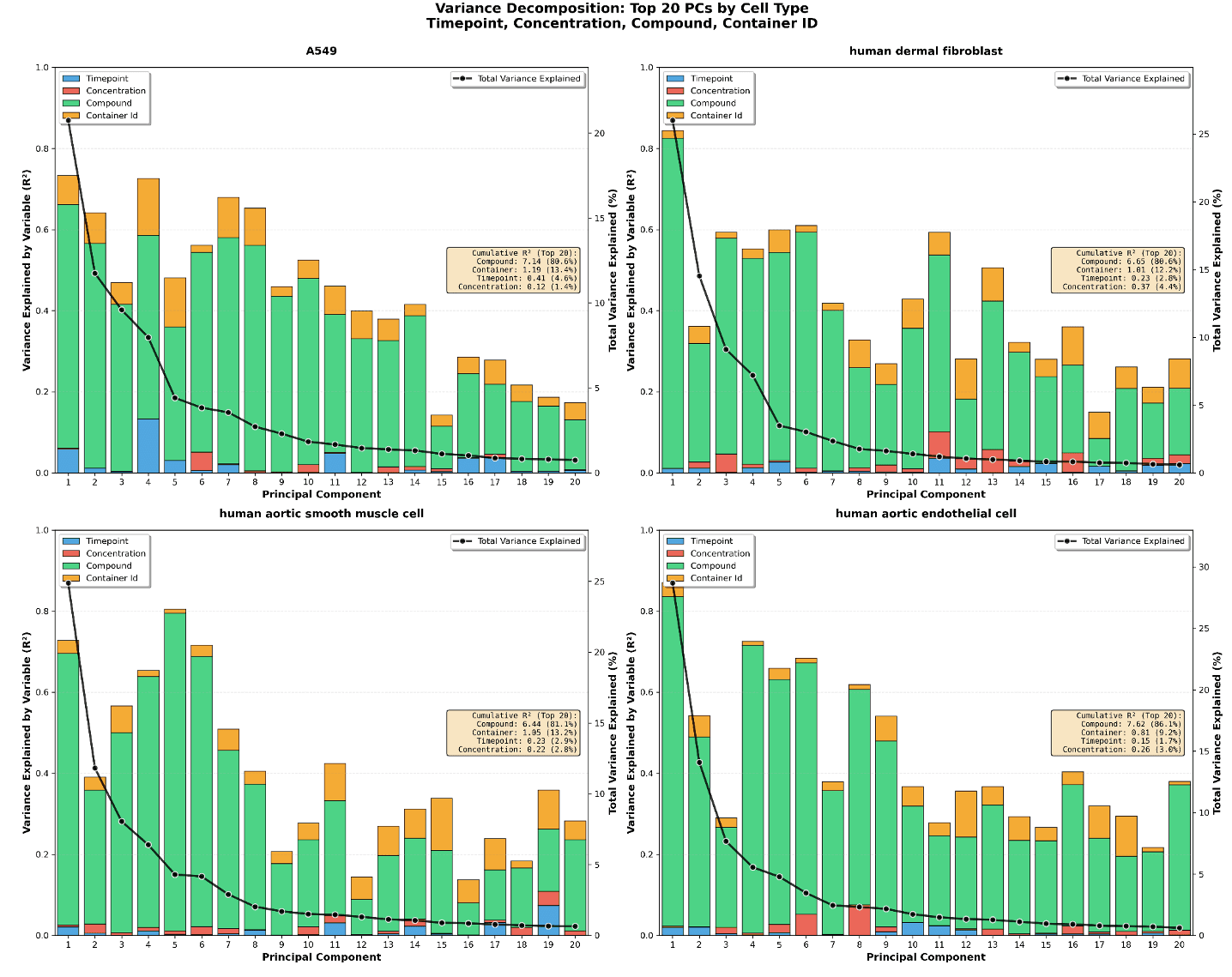
Figure 6. Top 20 PC’s for variance explained by timepoint, condition, concentration, compound, container_id, location.
Biomni summarized the data and also made the following interpretation of the results:
Endothelial cells: Best for high-throughput screening (highest compound sensitivity, lowest batch effects)
A549: Best for temporal studies (highest timepoint sensitivity)
Fibroblasts: Best for dose-response studies (highest concentration sensitivity)
Smooth Muscle: Unique PC structure (compound drives PC5 instead of PC1)
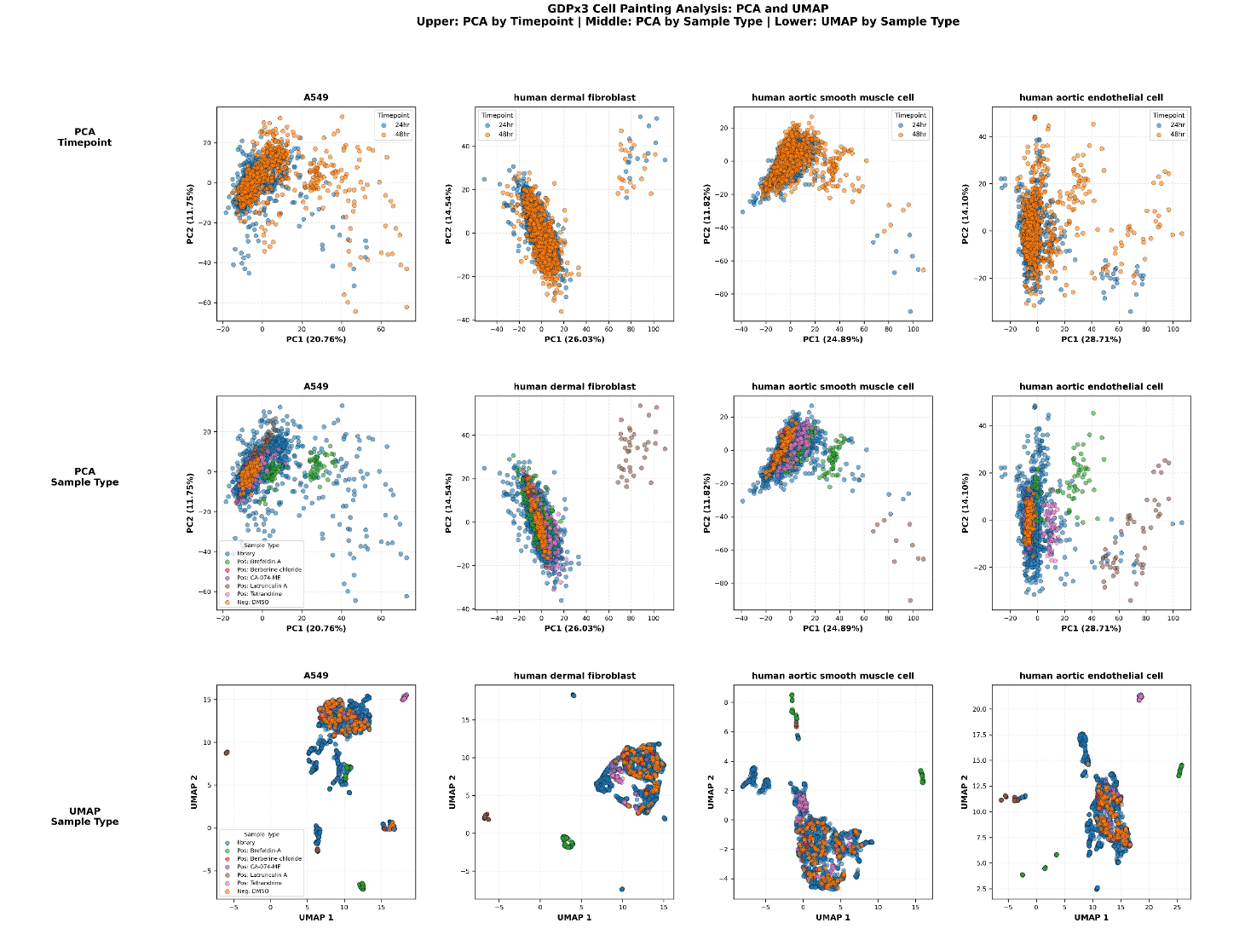
Figure 7. Top 20 PC’s for variance explained by timepoint, condition, concentration, compound, container_id, location
The PCA and UMAP plots are in-line with our expectations, where we observed compound effects to have the largest impact on PC variance. Additionally compounds LatrinculinA and BrefeldinA lead to the largest impact on the morphological data. LatrinculinA was toxic at all concentrations and for all cell types, whereas BrefeldinA had a clear effect on the ER morphology in line with its function as a disruptor of ER-Golgi trafficking.
Question 7:
“For each cell type using z-scored GDPx3_median_selected.csv values calculate the percentage of profiles that correlate with each other higher than DMSO samples in the same container. Generate a scatter plot with median pairwise spearman correlation between replicate profiles on the x-axis and -log10(pvalues) on the y axis.”
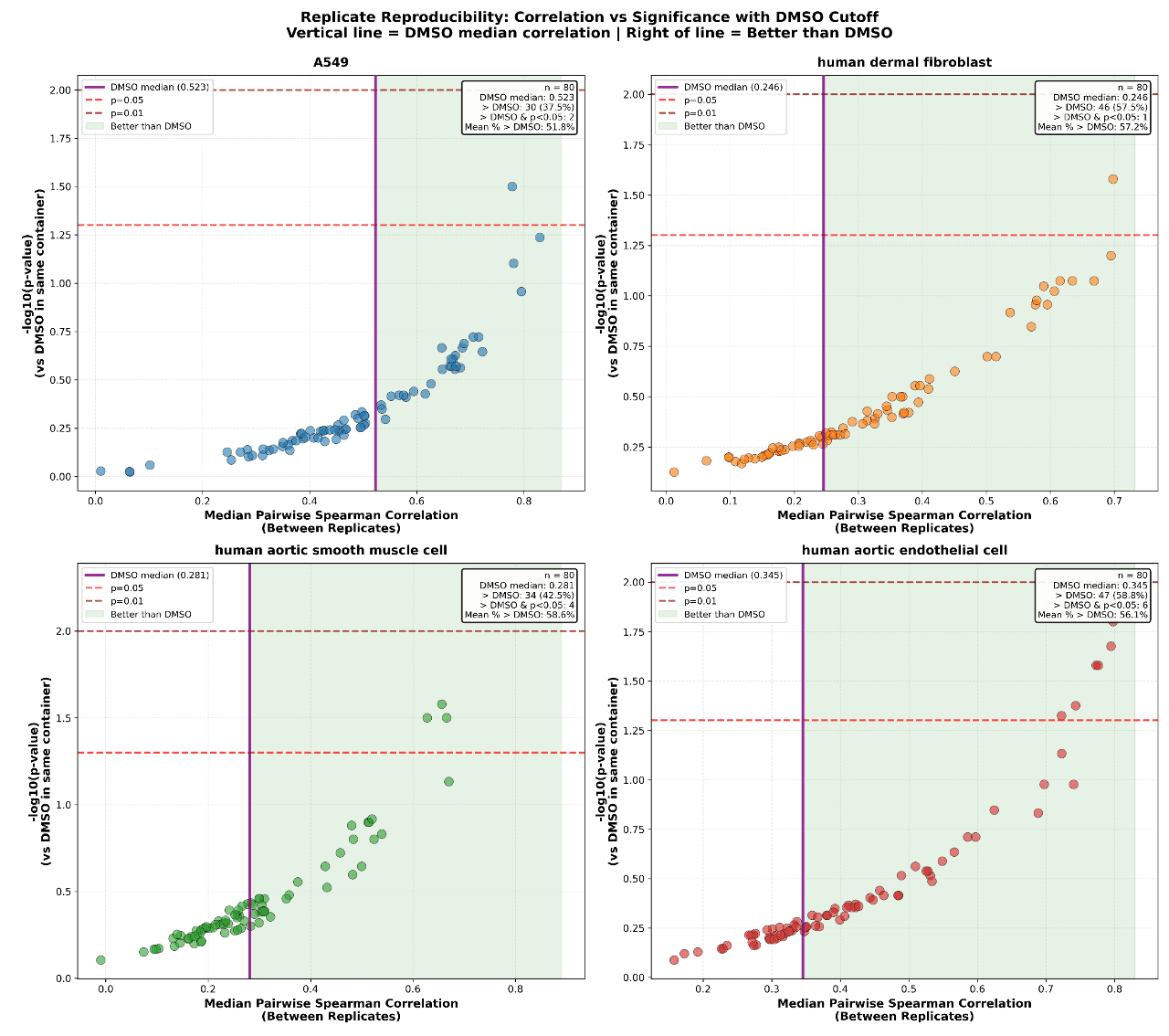
Figure 8. Percentage of profiles that correlate with each other higher than DMSO samples in the same container.
We observed significantly higher replicate correlation for DMSO in A549 cells whereas Dermal Fibroblasts had the lowest DMSO correlations. Consistent with these observations the identity of the compounds outperforming the DMSO treated cells are shown in Figure9 and are validated by our internal analysis. The compounds on the upper right quadrants have the highest pairwise correlation and significance relative to DMSO conditions in those cell types.
The Bottom Line
The Ginkgo X Biomni collaboration demonstrates how AI can fundamentally expedite interpretation and visualization of large scale datasets. Remarkably, what took Ginkgo's team of 2 people several weeks to produce was completed by the agent in just a few hours. This represents a dramatic leap in productivity while arriving at comparable conclusions and publication-ready figures. The Biomni tools are intuitive, easy to use, provide visibility and potential to update the plan of action at multiple steps in the analysis process.
By combining Ginkgo’s rich GDPx datasets with Biomni’s natural language-driven analysis capabilities, researchers can move from raw data to publication-ready insights. Together, we’re lowering the barrier to high-throughput data exploration and enabling scientists to uncover new biological hypotheses faster than ever before.
Experience It Yourself
Ready to explore? Visit https://biomni.phylo.bio/ and start asking questions about our GDPx datasets. You can explore these findings and generate your own hypotheses using natural language queries.
Ask questions like:
“Using GDPx2_GSEA-HallmarkGenes.csv, which pathways are uniquely activated in different cell types and by which compounds.”
"What cellular pathways are most affected by HDAC inhibitors in the GDPx2 datasets?"
"Which compounds in GDPx3 have similar mechanisms of action?"
The AI will analyze the data, generate visualizations, and provide scientific insights—all through natural language queries.