How the Biomni agent uses high-performance computing for biology
By Noah Lebovic, Visiting Member of Technical Staff at Phylo Zixin Huang, Member of Technical Staff at Phylo
Our goal is to give our biomedical AI agent, Biomni, the same capabilities as the researcher it’s working with. This post walks through how we give Biomni access to one of those capabilities: high-performance computing. This give it thousands of times more computational power than a laptop in a well-tested environment for running specialized computational biology software. This post gets more technical as it progresses. We’ll talk about why we built it, how it works, the architectural design, and the high-performance computing infrastructure itself.
When a researcher runs an analysis that’s too big for their laptop, they use powerful servers with more processing power and more storage. This type of computing is known as “high-performance computing” or “HPC”: powerful hardware, often with preconfigured scientific software, that supports research. Most research organizations maintain a resource like this for their staff.
Our goal is to give our biomedical AI agent, Biomni, the same capabilities as the researcher it’s working with. To do this, it uses a custom HPC environment when it needs more powerful servers or specialized preconfigured scientific software. This can give it thousands of times more computational power than a laptop in a well-tested environment for running specialized computational biology software.
For example, when performing structural biology tasks, Biomni uses the HPC environment to run tools like AlphaFold, Boltz, or Chai: tools that require a GPU and up to terabytes of files in their environment.
This post gets more technical as it progresses. We’ll talk about:
- Why we built it
- How it works
- Designing the interface between agent and HPC
- How we built the HPC infrastructure
Why we built the Biomni HPC, and what makes bio different
The Biomni agent runs inside of a sandbox that’s about as powerful as a laptop. That sandbox has many common bioinformatics tools preinstalled, so most agent sessions don’t need to use the Biomni HPC.
Some bioinformatics tools need more power. For example, a genome assembler that needs hundreds of gigabytes of RAM – or a structural biology workflow that requires a fleet of GPUs. The Biomni HPC helps us meet those resource demands without wastefully allocating them to every agent session.
Aside from meeting resource demands, we also use the Biomni HPC to isolate some specialized computational biology software. Some bioinformatics software can’t be installed through a package manager, like pip or conda, and environmental dependencies often conflict. In that case, it’s easier to just isolate the software to its own environment.
If you want to dive deeper into what makes bioinformatics workflows different, consider this post by Ben Siranosian.
An aside: reference databases
Reference databases are large files that a computational biology tool uses while analyzing data. Some tools, like AlphaFold, require terabytes of reference databases. AI folks can think of these as akin to model weights.
Downloading these files takes a long time – sometimes days. To make the HPC faster, we prestaged common reference databases on a high-speed shared filesystem.
How it works
The Biomni agent can use the HPC at any point while completing a task, and it knows which computational biology tools are set up in the HPC.
It doesn’t know the details of every tool, though – that would be too much information to keep in context. Instead, it knows only the basics: their names, and how to get more details. This type of progressive disclosure helps the agent maintain attention on longer tasks.
For example, Biomni can search for tools by name or category; “structural biology” returns detailed information on how to use tools like AlphaFold, Foldseek, Boltz-2, Sniffles, and STAR.
The detailed usage instructions the Biomni agent receives from the tool search are like a guide for humans. They include usage examples, descriptions of command-line arguments, and helpful notes for debugging. This almost always elicits reasonable usage from a frontier LLM, even if the computational biology tool wasn’t in a dataset or environment used when training the model.
Once the Biomni agent understands usage, it starts the tool using the HPC. Unlike a standard bioinformatics API, the agent writes the full command-line argument and sees the full output from the command it runs. Both of these help the model adapt to unexpected failures or niche usage.
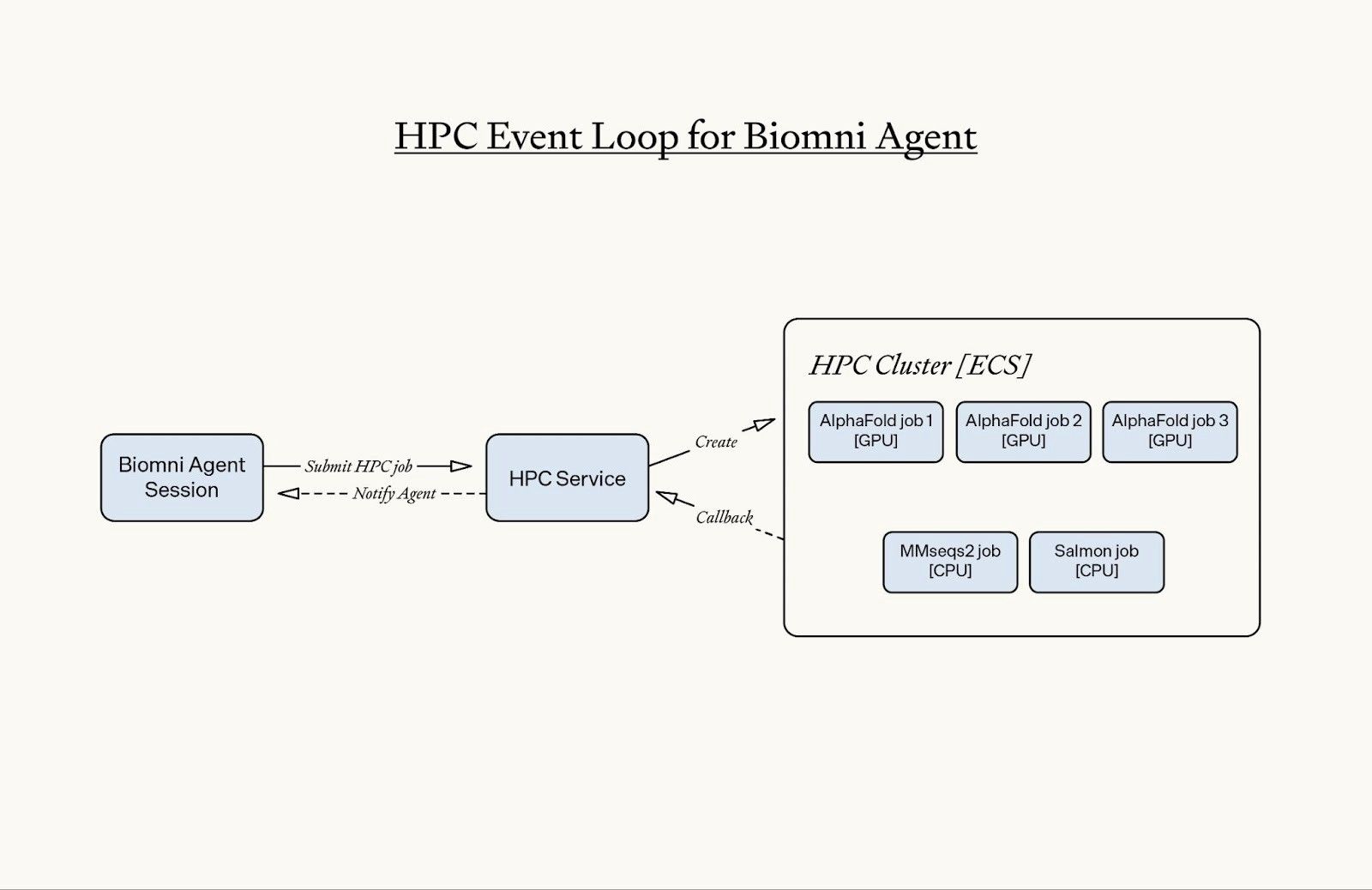
When the HPC job is submitted, it runs asynchronously in the background, and it schedules a `notify agent` callback. When the job finishes, the system automatically triggers this callback to inform the agent of the job’s status. If the agent is active, the notification is injected to the running session; if it’s idle, it wakes up the agent. The agent then resumes work from that point.
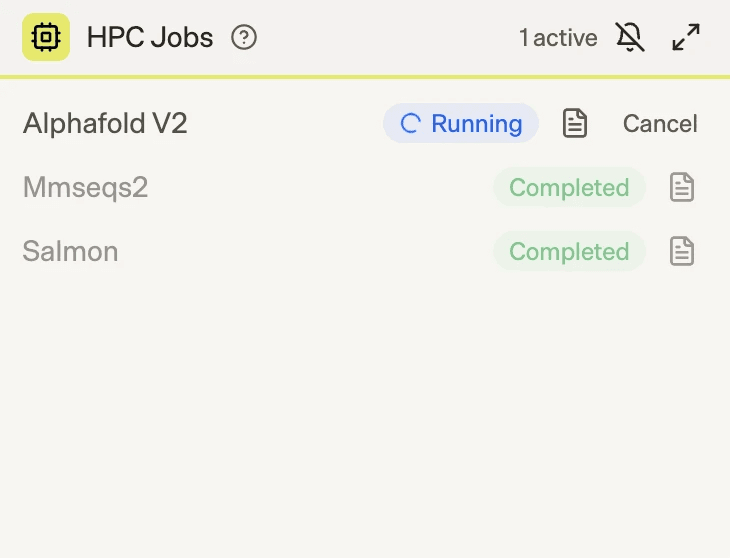
We also built an HPC Jobs widget that provides real-time visibility into running jobs. Researchers can monitor live job status, inspect logs at any time, and cancel jobs if they detect unexpected behavior. If a job is canceled, the callback mechanism immediately notifies the agent, which then continues the workflow accordingly.
Designing the interface between agent and HPC
The most interesting decision we faced was what level of flexibility to give the agent.
Concretely, we chose between:
- A rigid abstraction, where the LLM calls a rigid API with preimplemented and validated bioinformatics packages.
- A flexible abstraction, where the LLM runs arbitrary commands in preconfigured environments, but we limit its access to a set of constrained environments with usage instructions.
- No abstraction, where the LLM fully self-manages the infrastructure; this effectively grants the agent an AWS account of its own.
We decided on a flexible abstraction: one there’s a list of bioinformatics tools in preconfigured and tested environments, but the agent writes the shell commands that run in the environment. To help the agent write correct commands, each environment has a detailed usage guide.
In this paradigm, multiple commands can be run in the same HPC environment if the agent chooses to chain them together. For example, the output from the bioinformatics software kraken2 is often analyzed immediately by the bracken. Since the agent is writing the command, it can invoke both: kraken2 [...] && bracken [...].
The flexible abstraction’s resiliency to failure – even to issues outside of its scope, like out-of-memory errors – and its ability to support the long tail of use-cases make it more useful to the agent than a rigid API.
How we think about the value produced by different abstractions as we progress towards powerful AI.
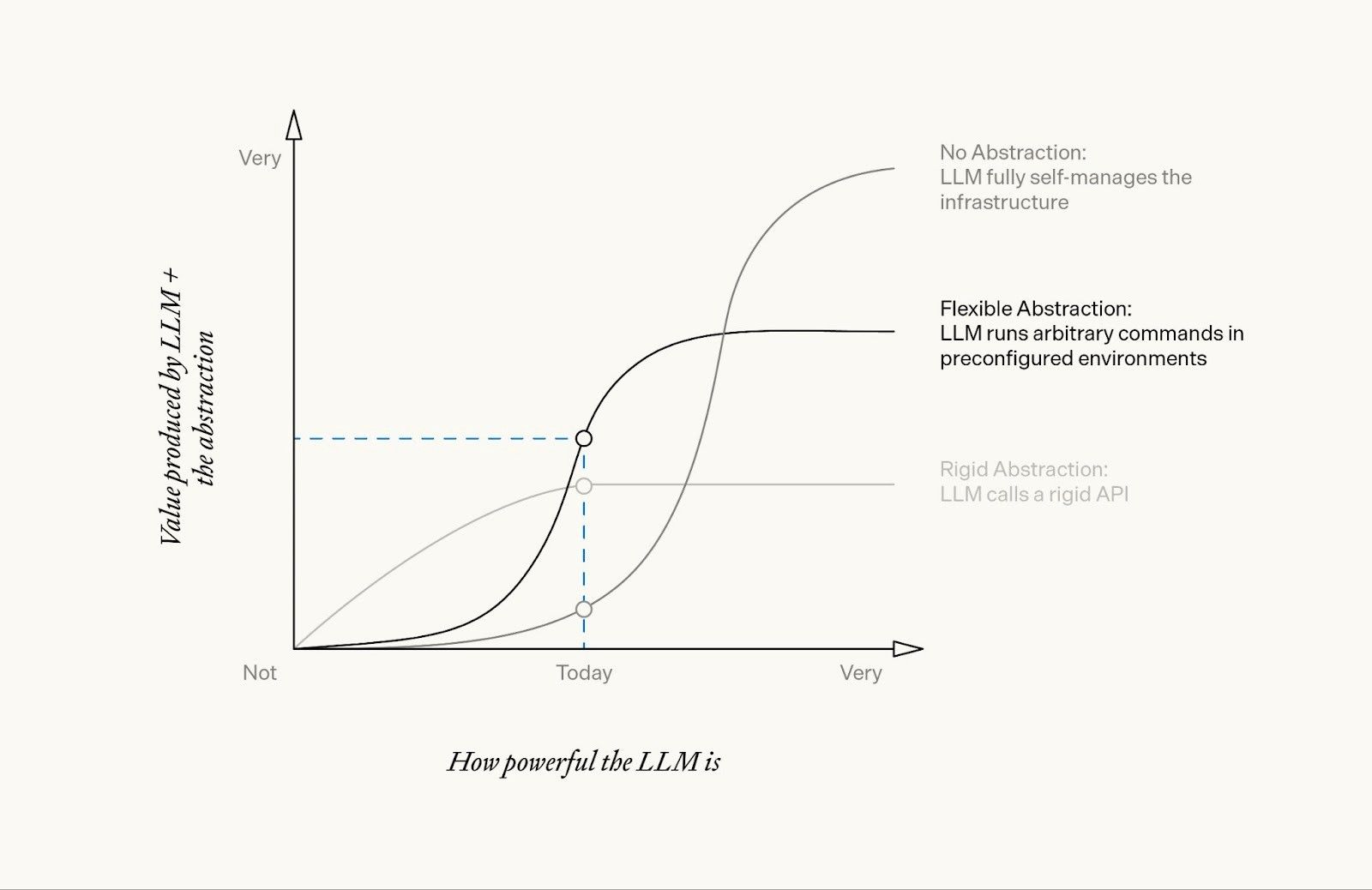
We expect that this flexible abstraction will continue to become more valuable as LLMs become more powerful, up until the point that the agent is able to effectively self-manage more complex infrastructure and its own environments.
Ultimately, the right abstraction depends on how powerful the AI is. While models from a year ago struggled with flexibility and needed rigid APIs, the current generation of models like Opus 4.6 or GPT 5.3 accomplish more when given a bit of flexibility.
The API
Once we’d decided on the proper level of abstraction, it was time to build the API. To do this, we designed an API to service six programmatic tools:
- Tool search
- Submit an HPC job
- Check a job’s status
- View a job’s logs
- Get results of a job
- Cancel an active job
For the purpose of this post, we’ll focus on just two: the tool search tool and submitting an HPC job.
Tool search
Under the hood, the tool search tool calls an API which searches a registry of tested tools. When Biomni searches for tools, a GET request is sent to the HPC API’s /v1/tools:
// curl -X GET "https://hpc.example.com/v1/tools?q=antibody" \
// -H "X-API-Key: secret-key-12345"
response = {
"type": "tool_search",
"query": "antibody",
"results": [
{
"type": "tool",
"id": "immunebuilder",
"name": "ImmuneBuilder",
"description": "Fast antibody/nanobody/TCR structure prediction...",
"usage": "Detailed usage instructions...",
"match_score": 95
},
}
],
[...]
}When Biomni receives the results, it knows to progressively parse them. The usage field can be particularly token-intensive.
Submit an HPC job
Biomni then submits a job, which – opaquely to the agent – sends a POST request to the HPC API’s /v1/jobs to spin up job:
// curl -X POST "https://hpc.example.com/v1/jobs" \
// -H "X-API-Key: secret-key-12345" \
// -H "Content-Type: application/json" \
// -d '{
// "tool_id": "immunebuilder",
// "inputs": [
// {
// "filename": "antibody.fasta",
// "type": "base64",
// "content": "PkhlYXZ5X2NoYWluCkVWUUxWRVNHR0dMVlFQR0dTTFJMU0NBQVM..."
// }
// ],
// "command": "ABodyBuilder2 --fasta_file /input/antibody.fasta -o /output/antibody.pdb -v"
// }'
response = {
"job_id": "hpcjob_abc123",
"status": "downloading_inputs",
"created_at": "2025-11-13T12:34:56.789Z",
"tool_id": "immunebuilder"
"type": "job"
}In the request above, the value of command is written by the agent with the usage guide. This flexibility is what helps over a rigid API: if the job fails, the agent can check the logs and rewrite the command to either fix it or to debug the environment.
There are downsides to this API, too: invalid parameters take longer to fail, and it’s possible for the agent to use a combination of parameters that just doesn’t make sense. On balance, it still seems to work better than a rigid API because of its resiliency to failure and support of niche tool uses that would be blocked by default in a rigid API.
Adding an HPC event loop for the Biomni agent to scale
As usage of the tool started scaling, we built two features to keep the HPC responsive and reliable. Two recurring issues that drove these features:
- The agent stopped and waited for HPC jobs to complete, which blocked the session from progressing.
- Some HPC jobs run for hours or days. These long jobs had a couple new failure modes:
- The agent might forget about the existence of the running HPC jobs after hours of user interaction and context compaction.
- The HPC job could exceed the agent sandbox timeout limits and disrupt the HPC lifecycle.
To fix this, we redesigned the HPC system to be async-first and introduced an agent notification callback mechanism.
With this architecture, the agent is notified when the HPC job completes. This removes blocking behavior from the agent’s execution loop and automatically reactivates an agent sandbox if needed. Because HPC jobs no longer block the session, researchers can continue interacting with Biomni while computational workloads run in the background – and they can leave the session without interrupting any ongoing jobs.
This shift to an async event-driven model helped us keep the HPC responsive and reliable as we scaled, and the user experience meshed better with real-world research workflows.
The infrastructure
On the AWS-based HPC executor, there are four key components:
- Tested container images (reproducible software environments)
- Pre-downloaded reference databases
- Staging for input and output files
- Infrastructure for HPC sandboxes
Tested container images
Every piece of bioinformatics software that’s available through the Biomni HPC has a corresponding image that’s tested for use with the software. We store these images on AWS Elastic Container Registry.
These images are larger than typical images used for web apps: they range from several hundred megabytes to dozens of gigabytes. Some of the images were custom-built by our team, and others were ported from amazing community resources like StaPH-B or Biocontainers and tested on our infrastructure.
To support maximum compatibility for different images, we have no dependency requirements inside of the image – there’s no requirement that the image contain command-line tools like aws, wget, curl, etc. Instead, we have separate Phylo-managed I/O containers that move input and output data to and from the environment.
Pre-downloaded reference databases
Many computational biology tools require reference databases, as we mentioned above. Small reference databases (e.g. less than 1 GB) can be included with the tool’s image, but larger reference databases (e.g. 6 TB) are well beyond the threshold that would be reasonable to include in an image; downloading the image alone would take half a day!
To solve this problem, we have a shared read-only filesystem that stores reference databases. We chose to use a Lustre filesystem, which is a distributed filesystem that’s often used in supercomputers. Importantly, Lustre is POSIX-compliant; many alternatives break when used with the gamut of bioinformatics tools, as some require operations that only POSIX-compliant filesystems provide.
This pattern lets us store massive reference databases in a shared read-only repository. Many HPC sandboxes can read from the filesystem at the same time, which is a well-supported pathway for Lustre. As the filesystem gets bigger, the throughput increases in tandem by “striping” files over many disks.
Staging for input and output files
Input and output files are shared with the Biomni HPC via reference (e.g. an S3 URI) or through a base64 encoded string directly in the request. When the Biomni HPC finishes a job, it transiently stores the outputs in an S3 bucket for retrieval by the API and the agent.
Infrastructure for HPC sandboxes
We use AWS ECS to manage the container lifecycle, with the containers themselves deployed to EC2 instances which we manage.
When Biomni submits a job to the HPC, our internal API matches the request with an appropriately resourced sandbox. Sometimes, this means a GPU-based instance, and other times it might be a large CPU-based instance with hundreds of gigabytes of RAM.
If there’s a sudden increase in demand for the HPC, the request is placed in a queue while the Biomni HPC provisions more resources. We dynamically provision more servers as needed, unlike the fixed cluster of servers that’s typical in an academic HPC.
Conclusion
Biomni uses its HPC tool for access to the same computational resources a researcher would use: powerful hardware, specialized software, and massive reference databases. By using a flexible abstraction – where the agent writes shell commands in preconfigured environments rather than calling rigid APIs – we’ve found a balance that works well with current frontier LLMs while leaving room to grow as the models improve.
We’re continuing to develop Biomni and its ecosystem to expand its capabilities. If this sounds interesting to you, consider joining us at phylo.bio/careers or try out Biomni at biomni.phylo.bio.